Bildquelle: Helena Klementova
Sichelzellkrankheit und (Beta-) Thalassämie gehören laut der Weltgesundheitsorganisation (WHO) zu den weltweit häufigsten Erbkrankheiten. In Deutschland treten sie bislang äußerst selten auf – ein Grund, weshalb auch betroffene Jugendliche schlecht über die Krankheitsbilder informiert sind. Forscher der Charité wollen das jetzt ändern und entwickelten eine App für die jungen Patienten.
Alle zwei Wochen eine Spritze in den Unterarm. Dann ein bis zwei Stunden warten, so lange bis die Bluttransfusion abgeschlossen ist. Und das nicht nur für einige Monate, sondern ein Leben lang – so oder ähnlich kann es Menschen gehen, die unter Sichelzellkrankheit oder Thalassämie leiden, zwei besonderen Formen der Blutarmut. Wie viele Menschen in Deutschland von diesen Erkrankungen betroffen sind, ist nicht bekannt. Schätzungen gehen von 3.000 bis 4.000 Fällen aus. Zahlen, die im Verhältnis zu anderen endokrinen Erkrankungen wie beispielsweise Diabetes Typ 1 kaum erschrecken. Sichelzell- und Thalassämie sind Exoten-, keine Volkskrankheiten. So sind auch viele Betroffene schlecht über ihre Erkrankung informiert. „Gerade unsere jugendlichen Patienten wissen oft nicht gut Bescheid“, erzählt Stephan Lobitz von der Klinik für Pädiatrie mit Schwerpunkt Onkologie und Hämatologie der Charité Universitätsmedizin Berlin. Nur wenige seien miteinander vernetzt und nutzten das Wissen oder die Hilfe der Community. Im Rahmen des Projektes „ePatients“ wollen Lobitz und sein Team jetzt eine App entwickeln – sie soll die jungen Patienten dazu motivieren, sich aktiv mit ihrer Erkrankung auseinanderzusetzten.
Erkrankungen des Blutes
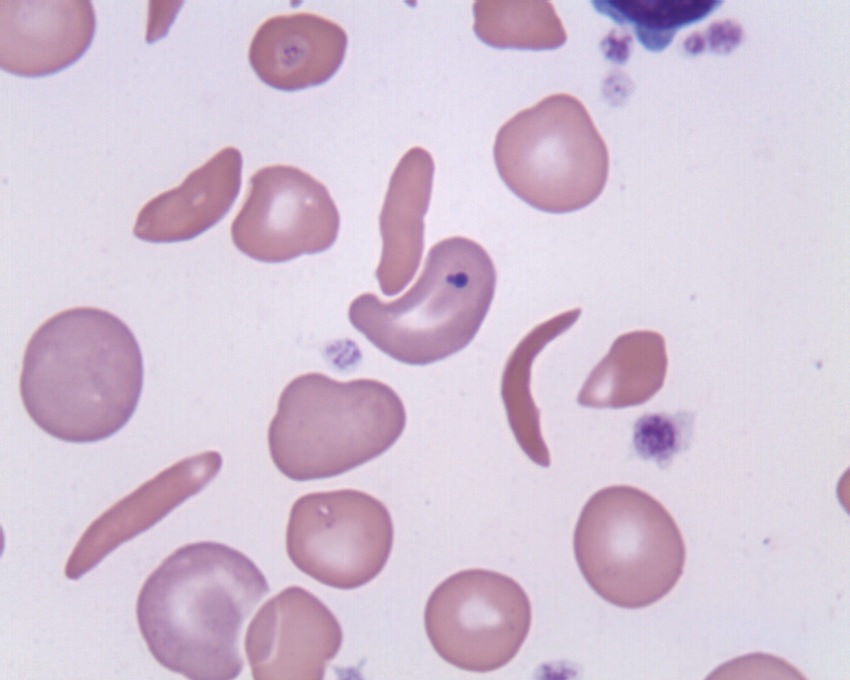
Sichelzellkrankheit wie auch Thalassämie sind zwei schwere Erbkrankheiten. In beiden Fällen wird der Körper der Betroffenen nicht ausreichend mit Sauerstoff versorgt. Der Grund ist ein Defekt der roten Blutkörperchen. Während es bei der Thalassämie dauerhaft an funktionstüchtigen roten Blutkörperchen mangelt, ist das Problem bei der Sichelzellkrankheit ein abnormes Hämoglobin (roter Blutfarbstoff). Nach der Abgabe des Sauerstoffs an die Organe des Körpers verformen sich die roten Blutkörperchen zu sichelförmigen Gebilden, die miteinander verklumpen, an Elastizität verlieren und in den Blutgefäßen stecken bleiben. Die Folge sind Gefäßverschlüsse, Blutarmut sowie das Risiko eines Infarktes. Medikamente und Bluttransfusionen können die Beschwerden zwar lindern; heilen lassen sich die Erbkrankheiten jedoch nur durch die Transplantation von Stammzellen. Wird die Blutarmut nicht rechtzeitig erkannt, kann sie bereits im frühen Kindesalter zum Tod führen.
Am häufigsten treten Sichelzell- und Thalassämie in den Ländern rund um das Mittelmeer und in Westafrika auf. Dass sich die Krankheiten gerade in diesen Gebieten durchsetzten, hat eine auf den ersten Blick überraschende Ursache: die Träger des Gendefekt sterben seltener an Malaria – eine Tropenkrankheit, die im Mittelmeerraum besonders häufig vorkommt. Sie sind zwar nicht immun, doch verläuft bei ihnen die Infektion weniger tödlich.
Neugeborenenscreening
Um Sichelzellanämie und Thalassämie rechtzeitig zu diagnostizieren und die Betroffenen optimal medizinisch versorgen zu können, führen Länder wie England, Frankreich, die Niederlande oder die USA Neugeborenen-Screenings durch. Die Leitlinien der deutschen Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) empfehlen ein solches Screening auch für Deutschland – bislang jedoch ohne Erfolg.
Mittelmeeranämie
Gerade Zypern galt lange Zeit als Malaria-Nest. Erst als Zypern 1925 britische Kronkolonie wurde, ließ sich die Fieberkrankheit zurückdrängen. Die Briten versprühten auf der gesamten Insel das Insektizid Dichlordiphenyltrichlorethan (DDT), wodurch sie die Mücke als Überträger nahezu ausrotteten. 1948 war die Insel malariafrei – doch die Thalassämie blieb. Aufgrund der mangelnden medizinischen Versorgung hatte fast jede Großfamilie Zyperns ein bis zwei Kindstote wegen der Blutarmut zu beklagen. Seit in den Achtzigerjahren Gentests und Pränataldiagnostik aufkamen und sich verbreiteten gehört das Screening auf Thalassämie im griechischen Teil der Insel zur Routine – eine Routine, die nicht unumstritten ist und viele Abtreibungen zur Folge hatte. Selbst die zyprisch-orthodoxe Kirche verlangt seit 1983 eine Art Screening-Zertifikat, wenn sich ein Paar trauen lassen möchte. Braut und Bräutigam sollen wissen, worauf sie sich eventuell einlassen. Auf Zypern braucht niemand eine App, die den Betroffenen erklärt, was solch eine Blutarmut eigentlich bedeutet.
Unkenntnis in Deutschland
In Deutschland sieht die Lage anders aus. Sichelzellkrankheit und Mittelmeeranämie gelten hier nach wie vor als Exotenkrankheiten. Malaria kam auf dem europäischen Festland nicht vor und so bot auch der Gendefekt keinen Überlebensvorteil. Erst durch Migration sind Sichelzell- und Thalassämie auch in Ländern wie Deutschland, England oder den Niederlanden angekommen.
Die App

© apprime GmbH
„In der Entwicklung der App arbeiten wir so eng wie möglich mit den Kindern und Jugendlichen zusammen “, erklärt Lobitz. Auch die Fragebögen wurden Schritt für Schritt gemeinsam entwickelt. Und die Ergebnisse bestätigen vorab formulierte Vermutungen: Bis zu 70 Prozent der Befragten bewerten „Tipps für Krisenfälle“ wie auch „Informationen zur Krankheit“ als die wichtigsten Funktionen der App. Wichtig sei den Nutzern aber auch der Wunsch nach Vernetzung mit anderen Betroffenen (Community-Funktion) sowie eine Erinnerungsfunktion zur Einnahme von Medikamenten und für die Vorsorgetermine. „Gerade in der Pubertät haben Jugendliche anderes zu tun, als sich nur mit ihrer Krankheit zu beschäftigen“, meint Lobitz, „da wird der Gang zum Arzt schon mal vergessen.“
Noch ist die App nur ein Entwurf. Bei den Jugendlichen kam die Idee jedoch gut an. Lobitz und sein Team wollen jetzt am Ball bleiben und hoffen, noch in diesem Jahr mit einer ersten Testversion der App starten zu können.
Information: Nachwuchsforschungspreis
Für die Entwicklung Entwicklung ihres Neugeborenenscreenings wurden Stephan Lobitz und sein Team am 26. Februar 2015 mit dem Anerkennungspreis für Nachwuchswissenschaftler im Rahmen des Eva Luise Köhler Nachwuchsforschungspreis für seltene Erkrankungen ausgezeichnet. Der Preis wird jährlich von der Eva Luise und Horst Köhler Stiftung in enger Kooperation mit der Allianz Chronischer Seltener Erkrankungen (ACHSE) e.V. vergeben.